Angelmanin oireyhtymä
Lisa Vogel opiskeli osastojournalismia keskittyen lääketieteeseen ja biotieteisiin Ansbachin yliopistossa ja syvensi journalistista tietämystään multimediatiedon ja viestinnän maisteriksi. Tätä seurasi harjoittelu -toimituksessa. Syyskuusta 2020 lähtien hän on kirjoittanut freelance -toimittajanaille.
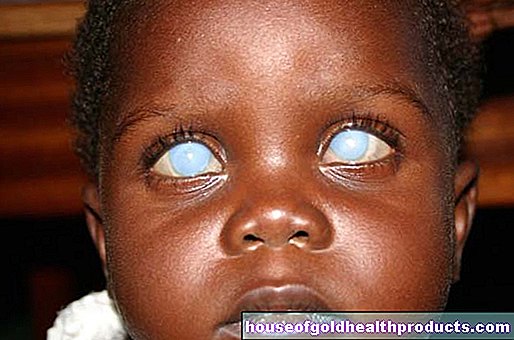
Lisää viestejä käyttäjältä Lisa Vogel Lääketieteelliset toimittajat tarkistavat kaiken -sisällön.Angelmanin oireyhtymä (Happy Puppet Syndrome) on harvinainen, geneettinen sairaus. Se ilmenee muun muassa henkisten ja fyysisten rajoitusten, kehityshäiriöiden (erityisesti kielen) ja yliaktiivisuuden kautta. Silmiinpistävää on nuken kaltainen ulkonäkö ja kärsivien onnellinen ilme. Lue lisää harvinaisesta sairaudesta!
Tämän taudin ICD -koodit: ICD -koodit ovat kansainvälisesti tunnustettuja koodeja lääketieteellisille diagnooseille. Ne löytyvät esimerkiksi lääkärin kirjeistä tai työkyvyttömyystodistuksista. Q93

Lyhyt katsaus
- Mikä on Angelmanin oireyhtymä? Harvinainen, geneettinen sairaus, joka ilmenee lapsen kehityksen henkisten ja fyysisten rajoitusten kautta
- Oireet: nuken kaltaiset kasvonpiirteet, kehityshäiriö, heikentynyt koordinaatio, kielitaidon puuttuminen tai ei lainkaan, älykkyyden heikkeneminen, kohtaukset, perusteeton nauru, naurut, liiallinen kuolaaminen, iloinen heilutus
- Syyt: geneettinen vika kromosomissa 15
- Diagnoosi: muun muassa keskustelu, fyysinen tutkimus, geneettiset tutkimukset
- Hoito: Kausaaliterapiaa ei ole saatavilla; Tukeva esim. Fysioterapia, puheterapia, toimintaterapia; mahdollisesti lääkitys oireiden lievittämiseksi (esim. kohtausten yhteydessä)
- Ennuste: normaali elinajanodote; itsenäinen elämä ei ole mahdollista
Angelmanin oireyhtymä: määritelmä
Angelmanin oireyhtymä (AS) johtuu kromosomin 15 geneettisestä viasta. Tämä vika häiritsee kärsivien fyysistä ja henkistä kehitystä. Puheen kehityshäiriöt, motorinen turvattomuus ja onnelliset kasvot ovat Angelmanin oireyhtymän näkyvimpiä oireita.
Nimi "Angelmanin oireyhtymä" tulee taudin keksijältä, englantilaiselta lastenlääkäri Harry Angelmanilta. Vuonna 1965 hän vertasi kliinisiä kuvia kolmesta lapsesta, joilla oli nuken kaltaisia kasvoja. Lapset nauroivat paljon ja tekivät nykiviä liikkeitä - kuten marionetteja Tämä johti englantilaiseen nimeen "Happy Puppet Syndrome" (onnellinen nukke).
Angelmanin oireyhtymää esiintyy molemmilla sukupuolilla. Taudin riski on noin 1: 20 000. Tämä tekee oireyhtymästä harvinaisen sairauden.
Angelmanin oireyhtymä: oireet
Angelmanin oireyhtymää sairastavat lapset ovat syntyessään normaaleja. Motoriset ja kognitiiviset kehityshäiriöt tulevat yhä havaittavammiksi vauvana ja varhaislapsuudessa. Geneettisen häiriön piirteitä ovat:
- viivästynyt moottorin kehitys
- heikentynyt koordinaatio
- usein ei lainkaan tai tuskin lainkaan kielen kehitystä
- älykkyys laski
- hyperaktiivinen, kiihkeä käyttäytyminen
- perusteetonta naurua
- Sopii nauruun
- iloisia eleitä (esim. heiluttaa käsiä)
- liiallinen kuolaaminen
- usein ulos kielestä
Joillakin lapsilla, joilla on Angelmanin oireyhtymä, on myös:
- Mikrokefalia (epänormaalin pieni pää) - ei syntyessään, mutta kun se kehittyy
- Kouristukset
- Muutokset aivojen sähköisessä toiminnassa
- erittäin vaalea iho ja silmät pigmentaation vähenemisen vuoksi (hypopigmentaatio)
- Squint (strabismus)
Angelmanin oireyhtymä: syyt
Angelmanin oireyhtymän syy on kromosomin 15 geneettinen vika: UBE3A -geenin toiminta on heikentynyt sairastuneilla. Tämä geeni tuottaa normaalisti entsyymin, joka osallistuu vaurioituneiden tai tarpeettomien proteiinien hajottamiseen soluissa. Se auttaa solua toimimaan normaalisti.
UBE3A -geeni sijaitsee kromosomialueella 15q11q13. Siellä geenit ovat alttiina niin kutsutulle "genomiselle jäljenjäljelle". Tämä tarkoittaa, että ne ovat aktiivisia vain yhdessä vanhempien kromosomeista (kehosoluissamme on kaksi kopiota jokaisesta kromosomista - yksi äidiltä ja toinen isältä). Tätä säätelee kemiallinen prosessi - metylaatio: tietyissä kohdissa kiinnittyneet metyyliryhmät voivat kytkeä geenin päälle tai pois päältä.
Geeni on aktiivinen molemmissa kromosomeissa lukuisissa kehon soluissa, mutta ei aivojen hermosoluissa: monilla siellä olevilla ihmisillä UBE3A -geeni isän kromosomissa 15 kytketään pois päältä painatuksella. Tämän seurauksena UBE3A on aktiivinen vain äidin kromosomissa 15 aivoissa. Tämä tarkoittaa myös: Jos äidin geenikopio osoittaa virheen, sitä ei voida korvata käyttämättömällä isän geenikopiolla. Ja juuri tämä yhdistelmä esiintyy Angelmanin oireyhtymässä: isän geenisegmentti on kytketty pois päältä, äidin geenisegmentti on viallinen.
Taustalla oleva geneettinen virhe voi olla erityyppinen:
- Poisto: Noin 75 prosentilla kaikista Angelmanin oireyhtymästä kärsivistä ihmisistä puuttuu asianmukainen alue 15q11q13, jossa on UBE3A -geeni äidin kromosomissa 15. Koska vastaava isän kromosomin 15 alue on "sammutettu" painamalla, keho voi käyttää entsyymiä, jonka rakennesuunnitelma on tallennettu UBE3A -sukupolveen, älä luo.
- Mutaatio UBE3A -geenissä: Spontaanisti tapahtuva geenimuutos aiheuttaa sen sisältämien tietojen menettämisen. Tämä pätee viidestä kymmeneen prosenttiin ihmisistä, joilla on Angelmanin oireyhtymä. Noin joka viides tapaus esiintyy perinnöllisessä mutaatiossa: täällä äiti kantaa jo geneettisen muutoksen isänsä kromosomissa.
- Kaksi isän kromosomia 15: sairastunut perii molemmat kromosomit 15 isältään, ei yhtään äidiltä (lääketieteellisesti kutsutaan "isän yksipuoleiseksi disomiksi 15"). Joten aktiivista UBE3A -geeniä ei ole. Tämä koskee noin 1-2 prosenttia kaikista Angelmanin oireyhtymäpotilaista.
- Painovirhe: UBE3A -geeni äidin kromosomissa 15 - kuten isän kromosomissa 15 - kytketään pois päältä tulostamalla. Lisäksi tietty osa kromosomista voi puuttua (poistaminen). Painovirhe löytyy yhdestä neljään prosenttiin Angelmanin oireyhtymän tapauksista.
Loput 10-15 prosenttia tapauksista Angelmanin oireyhtymän syy on tuntematon. Muuten: jos äidin geeni on kytketty pois päältä ja isän geeni on viallinen, lapset kärsivät ns. Prader-Willi-oireyhtymästä.
Onko Angelmanin oireyhtymä perinnöllinen?
Yleensä Angelmanin oireyhtymän uusiutumisriski on pieni. Tämä tarkoittaa sitä riskiä, että sairastuneen lapsen vanhemmat saavat muita lapsia, joilla on myös oireyhtymä. Yksittäistapauksissa tämä riski riippuu kuitenkin suurelta osin Angelmanin oireyhtymän taustalla olevasta geneettisestä viasta.
Esimerkiksi Angelmanin oireyhtymässä kahden isän kromosomin 15 (isän yksipuoleinen disomia 15) vuoksi se on alle prosentin. Toisaalta Angelmanin oireyhtymä, joka johtuu painovirheestä ja tietyn geenisegmentin menetyksestä (IC -deleetio), voi esiintyä puolessa kaikista tapauksista sisaruksella.
Tämä lisääntynyt riski on olemassa myös UBE3A -mutaation yhteydessä - edellyttäen, että äiti on jo geneettisen vian kantaja (noin 20 prosentissa kaikista mutaatiotapauksista). Tällaisissa tapauksissa äiti perii mutaation isältään. UBE3A muuttuu siksi äidin isän kromosomissa. Jos tämä on kytketty pois päältä, äiti ei huomaa mutaatiota. Hän voi kuitenkin siirtää kromosomin lapsilleen, missä se - silloin äidin kromosomina - voi aiheuttaa Angelmanin oireyhtymän.
Teoriassa Angelmanin oireyhtymäpotilaat voivat lisääntyä. Riippuen siitä, milloin syy -kromosomimuutokset tapahtuivat (esim. Jo sukusolujen kehityksen aikana tai heti hedelmöityksen jälkeen), riski on joskus erittäin suuri (jopa 100 prosenttia), että sairastuneet siirtävät taudin eteenpäin. Tästä ei kuitenkaan ole luotettavia tietoja. Syyskuussa 1999 oli yksittäistapaus: äiti, jolla oli Angelmanin oireyhtymä, välitti taudin täällä.
Angelmanin oireyhtymä: diagnoosi
Jos huomaat lapsellasi edellä kuvattuja oireita, lastenlääkäri on ensimmäinen yhteyshenkilö. Hän voi rajata mahdolliset syyt tarkemmin ja ohjata sinut ja lapsesi tarvittaessa erikoislääkärille.
anamneesi
Ensimmäinen vaihe diagnoosissa on perusteellinen sairaushistoria. Lääkäri kysyy sinulta erilaisia kysymyksiä lapsestasi, kuten:
- Mitä muutoksia huomasit lapsessasi?
- Onko lapsellasi fyysisiä valituksia?
- Voiko lapsesi istua?
- Kurottaako lapsesi esineitä?
- Puhuuko lapsesi?
- Onko lapsesi usein selvästi iloinen tai innoissaan?
- Nauraako lapsesi sopimattomissa tilanteissa, esimerkiksi silloin, kun hän kärsii?
Lääkärintarkastus
Tämän jälkeen suoritetaan fyysinen tutkimus. Lastenlääkäri testaa, missä määrin lapsi kehittää säännöllisesti motorisia ja henkisiä taitojaan. Tähän tarkoitukseen käytetään yksinkertaisia harjoituksia: Esimerkiksi lapsen tulee keskittyä leluihin tai kurkottaa valikoivasti rakennuspalikoon. Lääkäri kiinnittää huomiota myös lapsen ilmeisiin. Usein nauru, nuken kaltaiset piirteet ja kuolaaminen ovat kaikki merkkejä Angelmanin oireyhtymästä.
Jos fyysisen tarkastuksen jälkeen epäillään harvinaista sairautta, lääkäri ohjaa sinut neurologille ja ihmisen geneettiselle asiantuntijalle.
Geneettiset testit
Geneettinen testaus on tärkeä osa Angelmanin oireyhtymän diagnosointia. Lääkäri tarvitsee pienen näytteen lapsen soluista, jotka hän voi saada suun limakalvolta esimerkiksi ottamalla verinäytteen tai ottamalla vanupuikon. Näiden solujen tai asiaankuuluvan kromosomialueen 15q11q13 geneettistä materiaalia (DNA) tutkitaan sitten tarkemmin laboratoriossa.
Ensimmäisessä vaiheessa lääkärit kiinnittävät huomiota kromosomisegmentin metylaatiomalliin (metylaatioanalyysi / testi). Samojen näytteiden lisätestit (deleetioanalyysi, mutaatioanalyysi) auttavat selvittämään Angelmanin oireyhtymän syyn tarkemmin. Tätä varten voi olla tarpeen tutkia myös vanhempien geneettistä rakennetta. Tällä tavalla lääkärit voivat määrittää, onko siellä jo geneettinen vika.
Lisätutkimuksia
Jatkotutkimuksista on usein hyötyä. Esimerkiksi EEG: tä voidaan käyttää havaitsemaan muutoksia aivojen sähköisessä toiminnassa, kuten usein esiintyy Angelmanin oireyhtymässä. Silmälääkärin tutkimukset voidaan myös ilmoittaa.
Angelmanin oireyhtymä: Terapia
Angelmanin oireyhtymä on parantumaton - taudin geneettistä syytä ei voida korjata. Varhaisella puuttumisella voi kuitenkin olla positiivinen vaikutus kärsivien motoriseen ja henkiseen kehitykseen. Esimerkiksi säännöllinen fysioterapia on hyödyllinen. Se voi parantaa lasten motorisia taitoja, lievittää liikkumisrajoituksia ja auttaa estämään toissijaisia sairauksia, kuten selkärangan epämuodostumia. Lapset, joilla on Angelmanin oireyhtymä, hyötyvät myös muista hoitomenetelmistä, kuten toimintaterapiasta ja puheterapiasta.
Lisäksi jotkin Angelmanin oireyhtymään liittyvät oireet ja tilat saattavat vaatia kohdennettua hoitoa. Esimerkiksi kouristuslääkkeet (epilepsialääkkeet) auttavat kouristuksia vastaan ja rauhoittavat lääkkeet (rauhoittavat aineet) vaikeisiin unihäiriöihin.
Verein Angelman e.
Angelmanin oireyhtymä: sairauden kulku ja ennuste
Ensimmäinen elinvuosi
Vauvoilla, joilla on Angelmanin oireyhtymä, on todennäköisemmin ongelmia imetyksen, imemisen ja nielemisen kanssa. He usein työntävät kielensä ulos tai kuolaavat paljon. Lisäksi lapset, joilla on Angelmanin oireyhtymä, sylkevät usein (mikä usein sekoitetaan ruoka -intoleranssiin tai refluksitautiin). Usein oksentelu voi johtaa vaaralliseen laihtumiseen.
Kolmen tai kuuden kuukauden ikäisinä lapset, joilla on Angelmanin oireyhtymä, alkavat usein hymyillä. He nauravat ja gurgling paljon ja usein kielensä ulos näiden ilonpurkausten aikana.
Motorisen kehityksen viivästyminen on yleensä havaittavissa 6–12 kuukauden iässä: lapset eivät ryömi tai istu. Ylävartalon liikkeet ovat usein täriseviä. Tämä puolestaan vaikeuttaa istumista.
Murtavalla osalla sairastuneista on kohtauksia jo 12 kuukauden iässä.
Yksi tai kolme vuotta
Kolmen ensimmäisen elinvuoden aikana Angelmanin oireyhtymän kehityshäiriö tulee hyvin ilmeiseksi. Lapset kärsivät enemmän lievistä kohtauksista. Jotkut heistä ovat yliaktiivisia, yliaktiivisia ja aina liikkeellä. Monilla on taipumus laittaa kätensä tai leikkinsä suuhunsa koko ajan tai pistää kielensä usein ulos ja kuolata. Jos lapset ovat erityisen innoissaan, he usein nauravat liikaa ja heiluttavat ojennettuja käsiä.
Heikentynyt kielen kehitys ilmenee ensimmäistä kertaa tässä iässä. Lapset "hölmöilevät" itselleen tai huutavat ja kilisevät, mutta voivat kuitenkin lausua vain muutamia tai ei lainkaan helposti ymmärrettäviä sanoja ja yleensä käyttää niitä ilman kontekstia.Mutta he ymmärtävät kieltä ja siellä on myös sosiaalista vuorovaikutusta muiden ihmisten kanssa.
Murrosikä ja aikuisuus
Murrosikä on usein 3-5 vuotta myöhässä Angelmanin oireyhtymää sairastavilla lapsilla. Kuitenkin seksuaalinen kypsyys kehittyy normaalisti. Kielen kehittymistä ei edelleenkään tapahdu, jos kielen ymmärtäminen on usein läsnä. Aikuisen kohtauksia voidaan yleensä hoitaa hyvin lääkkeillä.
Angelmanin oireyhtymästä kärsivillä ihmisillä on normaali elinajanodote. Itsenäinen elämä ei ole mahdollista henkisten rajoitteiden vuoksi.
Tunnisteet: terveet jalat tcm kotihoito